Researchers from Centre for Quantum Geometry of Moduli Spaces (QGM), Department of Mathematics has in an interdisciplinary collaboration developed a mathematical method to analyze the three dimensional structure of proteins. The new method can be an important tool in the development of new drugs and is a step on the way to solve the long standing ‘Folding Problem’.
2014.12.18 |
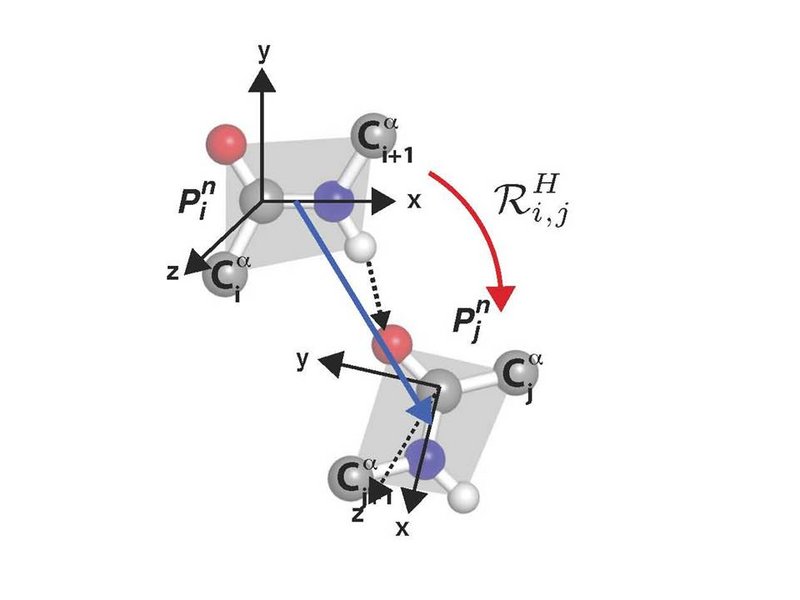
The new mathematical method offers a simple way to create a unifying overview of the structure elements of a protein, instead of having to investigate single structure aspect of the protein at a time. This is an important progress, as most proteins have a close correlation between its spatial structure and its function.
QGM Centre Director Jørgen Ellegaard Andersen has led the development of the method, which is done in close collaboration with people from different departments and centers at Aarhus University; Departments of Chemistry, Physics and Astronomy & Molecular Biology and Genetics, the three Danish National Research Foundation Centres of Excellence: Center for DNA Nanotecnology (CDNA), Center for Membrane Pumps in Cells and Disease (PUMPkin) & Center for Insoluble Protein Structures (inSPIN) as well as the Interdisciplinary Nanoscience Centre (iNANO).
Using methods from quantum field theory and quantum geometry a method for characterize each hydrogen bond of a protein by a rotation has been developed. The huge collection of data of more than 90.000 different proteins in the Protein Data Bank (PDB) has been used in the development of the method.
The content of the method is in brief that a hydrogen bond from a protein in PDB is chosen and it is determined, how the two connected peptide units in the hydrogen bond is placed in relation to each other. For each hydrogen bond, the whole molecule is rotated in such a way that the donating peptide unit in the chosen hydrogen bond is brought into standard position. The rotation affiliated with the chosen bond is the one which brings the first peptide unit in the standard position onto the other peptide unit that it is hydrogen bonded with. The rotation for all the hydrogen bonds in a protein gives a new dataset of vectors which is characteristic for the protein. These characteristics can be compared without any alignment needed. Further this characteristic rotation for each hydrogen bond constitutes a method for, on an equal footing, to sample and analyse secondary structures, turns, loops and unstructured region; something which so far have been processed individually.
The researchers have with this new mathematical model shown, that all hydrogen bonds for all proteins in PDB concentrates into 30 data clusters, taking up only 32% of the volume of all rotations. This pattern that the hydrogen bonds form is not a purely local but globally determined by the structure of proteins. It has been shaped through the evolution of the structures of proteins. This has been documented through a DFT analysis.
Contact: Jørgen Ellegaard Andersen
Read the scientific article - external link
See news article in ST Newsletter 'Rømer' December 2014 issue - AU link - (only in Danish)
News on DG website – external link
See article on Videnskab.dk - (only in Danish)
News on au.dk - (only in Danish)
